Computational materials science protocols
- Use
MolMod/exam_cif2xyz.mw, see MolMod package.
- Create CIF draft according to this template.
- Preprocess CIF draft:
- Generate entire molecule and move it (to the origin if possible).
- Change symmetry settings.
- If there are multiple symmetry-inequivalent molecules in the unit cell,
create separate CIF for each of them and process separately.
If molecules are naturally grouped (solute+solvent, charge-transfer pair),
they can be processed as a single meta-molecule.
All molecules must have identical and separated numbering.
- Make molecules connected.
- Reorder atoms for consistency with the rest of data.
In complex cases extract molecule,
reorder it with
MolMod/exam_Geometry.mw/Molecules: Partition molecule and identify fragments code,
and use the result as reference for reordering.
- Symmetrize molecule at fixed digits.
- Add missing or replace ill-positioned hydrogens with PM7-optmized ones using
MolMod/exam_Geometry.mw/Crystals: add missing hydrogens.
- Provide generators
g11,g12,...,g21,g22,... explicitly
and modify them in CIF to get the right unit cell
(gm1 generates m-th molecule, it should be a pure rotation if possible).
- Save XYZ (example) and keep also thus created CIF (example).
- Use
MolMod/exam_vas.mw, see MolMod package.
- Start with refined CIF (example) converted to XYZ (example).
- Each disorder group requires separate optimization.
- Use PBE-D3/PAW as optimal trade-off between accuracy and complexity.
- Use fast SCF iterators like ALGO=F or VF.
- If initial geometry is problematic preoptimize at fixed cell.
- Optimize cell+coordinates with k-spacing≈0.2 Å-1 (expected numerical accuracy is 0.1 meV/atom), ECUT=900 eV, and PREC=Accurate.
- Fix all used k-grids in
fopts.ini (example) according to the following guidelines:
- Note that for small-molecule organic semiconductors typical values
of K0 and V0 are 10 GPa and 10 Å3
resulting in 0.3 meV/atom energy difference at 3% volume change (example).
- k-spacing≈0.2 Å-1 is usually enough for smooth EoS with numerical accuracy better than 0.1 meV/atom.
- Use comparable-size k-grids for different polymorphs.
- Use the same number of k-points along topologically equivalent directions.
- MBD, highly dispersive bands, and flat PES might require k-spacing as low as 0.15 iAo.
- Gaussian smearing with default SIGMA=0.2 eV might be inaccurate for narrow-gap systems like hexacene with PBE functional or charge-transfer compounds.
- Set convergence criteria according to the following guidelines:
- Default EDIFF=1e-4 and tightened EDIFFG=1e-4 (0.1 meV) should give geometry with 1% accuracy and final gradients of the order of tens meV/Ao.
- To improve accuracy set EDIFFG=1e-5 and maybe EDIFF=1e-5.
- For numerically precise geometry optimization set EDIFF=1e-6 and EDIFFG=-1e-3 (1 meV/Ao).
- For large unit cells, accuracy can be increased gradually in the following order:
- Increase ECUT from 400.
- Increase PREC from normal to accurate.
- Increase k-grid from minimal reasonable (such as Γ-point only, adding more points for highly dispersive directions).
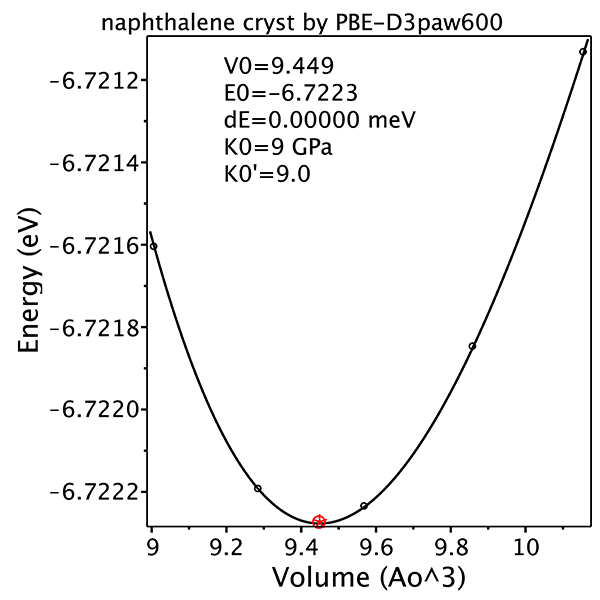
- If feasible, get final geometry by 5-point volume scan with 3% volume change step at ECUT=600
(example, if number of geo iterations exceed 30-40,
additional run is needed to converge all volume scan points with precision of fractions of meV/atom).
Otherwise reoptimize atomic positions at ECUT=400 (minimum for organics).
- Decrease k-spacing to 0.06-0.1 Å-1 to calculate final total energy and eDOS.
- Check consistency of all calculations by volume scan plot and EoS fit (especially K'0).
- Symmetrize or at least check symmetry at each run. Check that CIF is not corrupted. Check supersymmetry and symmetry breaking.
- If feasible calculate Γ-point phonons and elastic tensor to check the stability (example).
- Use
ITIC4F/dat/main.mw/Crystal
- It is assumed that all molecules are connected.
- Group molecules into translationally invariant sets.
- Determine cell translations using a reference atom and then fold cells.
- Superimpose molecules to inspect conformational disorder. It is assumed that the conjugated backbone is not conformationally disordered.
- Determine disorder groups for side chains:
1) Determine disordered part of side chains and type of disorder using both cartesian and internal coordinates.
2) Analyze disorder by histograms in proper coordinates.
3) Use cluster analysis (density-based such as DBSCAN and Gaussian distribution-based) to determine disorder groups.
4) Discard nonessential groups.
- Fix a disorder group and calculate average molecule.
Identify atoms whose average position is meaningless and determine their mean positions in internal coordinates.
In particular, aliphatic side chains often can be defined only by fixing C-C-C-C dihedrals to their most probable value
consistent with average position of the chain in cartesian coordinates.
- Check intramolecular symmetry and symmetrize.
- Determine unit cell origin, check crystal symmetry, and symmetrize.
- Optimize geometry with any light but accurate method to redetermine the space group.
- Use
QFT/Models/ECG/ECG.mw or MolMod/LocalizeMO.mw, see MolMod package.
- PBC are supported only at Gamma point (supercell approach).
- Fragmentize system, identify repeating fragments (monomers), and save to file following quantities:
1) fragments as
F=list(list(atom#));
2) bonds between fragments as B=list([atom#,atom#]);
3) terminal bonds as T=list([atom#,atom#]) with the 1st atom belonging to a fragment;
4) fragments to monomers map as f2m=list(monomer#);
5) monomers to fragments map as m2f=list(list(fragment#));
6) reference fragment for each monomer as m2fref=list(fragment#);
7) optionally monomer names as MN=list(string);
8) optionally symmetry information (translation period, asymmetric unit).
- Calculate MOs and save out,evl,evc,S (or evr).
- Select subspace of MOs for coarse-graining as accurate as possible,
though small number of irrelevant MOs can be discarded later during orbital localization.
- Prelocalize by geometry or other methods such as NBO or spatial localization,
so that you will get a list of pre-LMOs.
- Localize by projection onto pre-LMOs, filter out redundant MOs and pre-LMOs,
iterate until convergence (usually the second iteration is either final or meaningless).
Correct LMO signs by a sign function, or by reference orbitals, or manually, or applying a combination of these methods.
Save the obtained LMOs as evc,evr,h1e (optionally s1e) binary files and plain-text records containing:
1) copy of fragmentation information (see above);
2) basis set fragmentation as
K=list(list(basisfunction#));
3) LMOs to fragments map as o2f=list(fragment#);
4) fragments to LMOs map as f2o=list(list(LMO#));
5) list of coarse-grained MOs MOs=list(MO#);
6) localization method as method=string.
- Use
MolMod/_exam_Heff.mw, see MolMod package.
- Make sure that the crystal structure file contains information about symmetry and partitioning of the crystal into molecules:
SG=name, nm=number
- At the accuracy level of dimer approximation, it is usually enough to consider hydrogen-passivated pi-conjugated backbones instead of entire molecules.
- Load crystal structure and fragmentize molecules if needed,
then rationalize site positions and initialize FGS subpackage of FiniteGroups package.
- Step 1: Determine nearest neighbors (for molecules, not fragments) and save all symmetry unique dimers in xyz-file.
Normally only pi-conjugated atoms should be considered.
The cutoff distance of about 5~Ao can be used to screen out negligible wave-function overlaps (not for Forster energy transfer).
- Step 2: Perform required single point calculations (MOs or excited states)
for all translationally unique molecules, symmetry-unique dimers,
and optionally multimers (list of multimers should be provided by user).
For each calculation save out,evc,evr,evl files for frontier orbitals (let say, 8 MOs per band per monomer + 2 extra orbitals).
- Step 3: Fragmentize basis if needed (for each molecule) and save it as evc,evr,h1e files.
You will need reference LMOs to correct wave-function sign for each LMO.
- Step 4: Calculate transfer integrals and save Hamiltonian in this format.
For electrons and holes, a CG-basis is initialized either pre-calculated or as molecular HOMO/LUMO.
In the latter case the wave-function sign is corrected at this point.
The CG Hamiltonian is calculated by projection on CG-basis for all available multimers.
Then either mean (default) or most appropriate (user-defined) value is selected for evaluation of transfer integrals.
Small transfer integrals are discarded for noncontact pairs.
Finally, unfold symmetry-unique dimers and save the Hamiltonian in dat-file.
- Step 5: Print transfer integrals, calculate hopping amplitudes and band structure.
The report is saved to txt-file.
- Use
QFT/Models/TB.mw
- Create lattice geometry initialization file in this format supplemented with CIF.
- Create TB Hamiltonian initialization file in this format,
including list of symmetry-unique transfer integrals, visualized with CIF and picture.
- Save symbolic Hamiltonian in this format.
- Parameterize this TB Hamiltonian from supercell or cluster calculations, saving the result in the TB Hamiltonian file.
- Derive eigenvectors and eigenvalues at high-symmetry k-points and k-paths.
- Derive a minimal TB model and plot its band structure and DOS.
- If relevant, parameterize the TB model from high-symmetry k-points.
- Derive hopping amplitudes tensor and inverse mass tensors at selected eigenstates.
- Study convergence of TB models wrt number of parameters and supercell/cluster size.
{kind=link}